
TRAMETINIB NOVARTIS 0,05 mg-mL poudre pour solution buvable, boîte de 1 flacon de 90 ml
Retiré du marché le : 16/07/2025
Dernière révision : 01/08/2023
Taux de TVA : 0%
Laboratoire exploitant : NOVARTIS PHARMA
Source :

Gliome de bas grade
Trametinib 0,05 mg/mL est indiqué en association à Dabrafenib 10 mg comprimé dispersible dans le traitement des patients pédiatriques âgés de 1 an et plus atteints d'un gliome de bas grade (GBG) porteur d'une mutation BRAF V600E qui nécessitent un traitement par voie systémique.
Gliome de haut grade
Trametinib 0,05 mg/mL est indiqué en association à Dabrafenib 10 mg comprimé dispersible dans le traitement des patients pédiatrique âgés de 1 an et plus atteints d'un gliome de haut grade (GHG) porteur d'une mutation BRAF V600E qui ont reçu au moins un traitement antérieur par radiothérapie et/ou chimiothérapie.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
La solution buvable de 0,05 mg/mL est destinée à être utilisé en association à Dabrafenib 10 mg comprimé dispersible. Le RCP du dabrafenib en comprimés dispersibles doit être consulté avant l'initiation du traitement. Pour plus d'informations sur les mises en garde et les précautions d'emploi associées au traitement par dabrafenib, veuillez vous référer au RCP de dabrafenib comprimés dispersibles.
Les données d'efficacité disponibles d'un traitement par monothérapie (uniquement par dabrafenib ou trametinib), et notamment par trametinib seul, sont limitées.
Nouvelles tumeurs malignes
De nouvelles tumeurs malignes, cutanées ou non cutanées, peuvent survenir lorsque dabrafenib est utilisé en association au trametinib.
Tumeurs cutanées
Des tumeurs cutanées malignes telles que des carcinomes épidermoïdes cutanés (CEC) incluant des cas de kératoacanthome et de nouveaux mélanomes primitifs ont été observés chez les patients adultes traités par trametinib en association au dabrafenib (voir la rubrique Effets indésirables). Il est recommandé d'examiner la peau du patient avant l'initiation du traitement par trametinib puis mensuellement pendant le traitement et jusqu'à 6 mois après le traitement. Le contrôle de la peau doit continuer pendant 6 mois après la fin du traitement ou jusqu'à l'initiation d'un autre traitement antinéoplasique.
Les lésions de la peau suspectes doivent être prises en charge par une exérèse chirurgicale et ne nécessitent pas de modification du traitement. Les patients doivent être informés d'avertir immédiatement leur médecin en cas d'apparition d'une nouvelle lésion de la peau.
Tumeurs malignes non cutanées
Sur la base de son mécanisme d'action, dabrafenib pourrait majorer le risque de tumeurs malignes non cutanées en présence de mutations RAS. Veuillez vous référer au RCP de dabrafenib en comprimés dispersibles (rubrique Mises en garde spéciales et précautions d'emploi). Lorsqu'il est pris en association au dabrafenib, aucune adaptation posologique du trametinib n'est nécessaire pour des cancers ayant une mutation RAS.
Hémorragie
Des évènements hémorragiques ont été rapportés chez des patients adultes et pédiatriques prenant du trametinib en association au dabrafenib (voir la rubrique Effets indésirables). Des événements hémorragiques majeurs et des hémorragies d'issue fatale, sont survenus chez les patients adultes traités par trametinib et en association au dabrafenib. Le risque potentiel de ces évènements chez les patients présentant un taux de plaquettes bas (<75 000) n'a pas été établi étant donné que ces patients avaient été exclus des études cliniques. Le risque hémorragique peut être majoré par l'utilisation concomitante de médicaments antiplaquettaires ou anticoagulants. En cas d'hémorragie, les patients doivent être traités en fonction du tableau clinique.
Réduction de la fraction d'éjection du ventricule gauche / dysfonction ventriculaire gauche
Des cas de diminution de la FEVG ont été rapportés lors du traitement par trametinib en association au dabrafenib à la fois chez des patients adultes et pédiatriques (voir la rubrique Effets indésirables). Dans les essais cliniques chez les patients pédiatriques, le délai médian de survenue d'une diminution de la FEVG était d'environ 1 mois. Dans les essais cliniques chez les patients adultes, le délai médian de survenue d'une dysfonction ventriculaire gauche, d'une insuffisance cardiaque, ou d'une diminution de la FEVG était entre 2 et 5 mois.
Le trametinib doit être utilisé avec prudence chez les patients présentant une altération de la fonction ventriculaire gauche. Les patients ayant une dysfonction ventriculaire gauche, de stade II, III ou IV selon la classification NYHA, ayant présenté un syndrome coronarien aigu dans les 6 derniers mois, une arythmie non contrôlée cliniquement significative et une hypertension artérielle non contrôlée ont été exclus des essais cliniques ; la sécurité d'emploi dans cette population n'est donc pas connue. Chez tous les patients, la FEVG doit être évaluée avant l'instauration du traitement, un mois après l'instauration du traitement, puis environ tous les 3 mois durant tout le traitement (voir la rubrique Posologie et mode d'administration pour les informations concernant les adaptations posologiques).
Chez les patients traités par trametinib en association au dabrafenib, des cas de dysfonctionnements aigus, sévères du ventricule gauche dus à une myocardite ont été rapportés occasionnellement. Une récupération complète a été observée à l'arrêt du traitement. Les médecins doivent être vigilants quant à la possibilité de survenue d'une myocardite chez les patients qui développent pour la première fois ou aggravent des signes ou symptômes cardiaques.
Pyrexie
Des cas de fièvre ont été rapportés dans les essais cliniques chez des patients adultes et pédiatriques évaluant le trametinib (voir la rubrique Effets indésirables). L'incidence et la sévérité de la pyrexie ont été majorées avec le traitement en association (voir la rubrique Mises en garde spéciales et précautions d'emploi du RCP de dabrafenib en comprimés dispersibles). Chez les patients recevant trametinib en association au dabrafenib, la pyrexie peut être associée à des frissons sévères, une déshydratation et une hypotension qui, dans certains cas, peut conduire à une insuffisance rénale aiguë. Chez les patients pédiatriques qui ont reçu le trametinib en association au dabrafenib, le délai médian de survenue de la pyrexie était de 1,3 mois.
Le traitement avec le trametinib et le dabrafenib doit être interrompu si la température corporelle du patient est ≥ 38°C (voir la rubrique Propriétés pharmacodynamiques). En cas de récurrence, le traitement peut également être interrompu au premier symptôme de pyrexie. Un traitement par antipyrétiques tels que l'ibuprofène ou le paracétamol doit être instauré. L'utilisation de corticostéroïdes par voie orale doit être envisagée dans le cas où les antipyrétiques s'avèrent insuffisants. Les patients doivent faire l'objet d'une surveillance en vue de détecter tout signe ou symptôme évocateur d'infection. Le traitement peut être repris dès lors que la fièvre a disparu. Si la fièvre est associée à d'autres signes et symptômes sévères, le traitement doit être redémarrée à une dose réduite une fois l'épisode fébrile résolu et si l'état clinique du patient le permet (voir la rubrique Posologie et mode d'administration).
Modifications de la pression artérielle
Des hypertensions et des hypotensions ont été rapportées dans les essais cliniques avec le trametinib en association au dabrafenib (voir la rubrique Effets indésirables). La pression artérielle doit être mesurée à l'instauration du traitement et surveillée pendant le traitement et prise en charge par un traitement standard du contrôle de l'hypertension artérielle, si nécessaire.
Atteinte pulmonaire interstitielle/pneumopathie
Dans un essai de phase III chez les patients adultes, 2,4 % (5/211) des patients traités par trametinib en monothérapie, ont développé une atteinte pulmonaire interstitielle ou pneumopathie ; les 5 patients ont nécessité une hospitalisation. Le délai médian de survenue d'une atteinte pulmonaire interstitielle ou d'une pneumopathie a été de 160 jours (entre 60 et 172 jours). Dans les deux études des patients adultes traités par l'association de trametinib et dabrafenib, 1% des patients ont développé une pneumopathie ou une atteinte pulmonaire (voir la rubrique Effets indésirables).
Le traitement par trametinib doit être suspendu chez les patients avec une suspicion d'atteinte pulmonaire interstitielle ou pneumopathie, incluant des patients présentant de nouveaux signes et symptômes pulmonaires ou une progression de signes et symptômes préexistants, incluant une toux, une dyspnée, une hypoxie, un épanchement pleural ou des infiltrats pulmonaires, dans l'attente des résultats des investigations cliniques. Le trametinib doit être arrêté définitivement chez les patients ayant un diagnostic avéré d'atteinte pulmonaire interstitielle ou pneumopathie associée au traitement (voir la rubrique Posologie et mode d'administration). Le traitement par dabrafenib peut être poursuivi à la même dose.
Troubles visuels
Des troubles, associés à des perturbations visuelles, tels qu'un décollement de l'épithélium pigmentaire de la rétine ou une occlusion de la veine rétinienne peuvent survenir lors du traitement par trametinib. Des symptômes tels qu'une vision floue, une baisse de l'acuité visuelle et d'autres troubles visuels ont été rapportés dans les essais cliniques réalisés avec le trametinib chez l'adulte. Au cours des essais cliniques, des cas d'uvéite et d'iridocyclite ont aussi été signalés chez des patients adultes et pédiatriques traités par trametinib en association au dabrafenib.
Lors des essais cliniques pédiatriques il a été observé que les troubles visuels survenaient bien plus tardivement (délai médian de survenue > 3 mois) que les autres effets indésirables observés lors du traitement par trametinib en association au dabrafenib.
Le trametinib n'est pas recommandé chez les patients ayant des antécédents d'occlusion de la veine rétinienne. La sécurité d'emploi du trametinib n'a pas été établie chez des patients ayant des facteurs prédisposant à l'occlusion de la veine rétinienne, incluant un glaucome non contrôlé ou une hypertension oculaire, une hypertension artérielle non contrôlée, un diabète non contrôlé ou des antécédents de syndromes d'hyperviscosité ou hypercoagulabilité.
Un patient chez lequel survient un trouble de la vue, tel qu'une diminution de la vision centrale, une vision floue ou une perte de l'acuité visuelle durant le traitement par trametinib, devra être soumis rapidement à une évaluation ophtalmologique. Si un décollement de la rétine est diagnostiqué, les recommandations d'adaptation de posologie du Tableau 4 doivent être suivies (voir la rubrique Posologie et mode d'administration) ; si une uvéite est diagnostiquée, veuillez vous référer à la rubrique Mises en garde spéciales et précautions d'emploi du RCP de dabrafenib en comprimés dispersibles. Chez les patients pour lesquels une occlusion de la veine rétinienne est diagnostiquée, le traitement par trametinib doit être arrêté définitivement.
En cas de survenue d'une occlusion de la veine rétinienne ou d'un décollement de la rétine, aucune adaptation posologique de dabrafenib n'est nécessaire lorsqu'il est pris en association au trametinib. En cas de survenue d'une uvéite, aucune adaptation posologique de trametinib n'est nécessaire lorsqu'il est administré en association au dabrafenib.
Eruption cutanée
Des éruptions cutanées ont été observées chez environ 47 % des patients pédiatriques au cours des essais cliniques quand le trametinib est utilisé en association au dabrafenib (voir la rubrique Effets indésirables). Dans la majorité des cas, les éruptions étaient de grade 1 ou 2 et n'ont pas nécessité de réduction de posologie ou d'interruption du traitement.
Effets indésirables cutanés graves
Des cas d'effets indésirables cutanés graves incluant le syndrome de Stevens-Johnson, et la réaction d'hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (DRESS pour Drug Reaction with Eosinophilia and Systemic Symptoms), pouvant menacer le pronostic vital ou être fatals, ont été rapportés lors de traitement par l'association dabrafenib/trametinib chez les patients adultes. Avant d'initier le traitement, les patients doivent être informés des signes et symptômes des réactions cutanées et être étroitement surveillés. Si des signes et symptômes évocateurs d'une réaction cutanée grave apparaissent, le dabrafenib et le trametinib doivent être arrêtés.
Rhabdomyolyse
Des cas de rhabdomyolyse ont été rapportés chez des patients adultes recevant du trametinib. Dans certains cas, les patients ont pu poursuivre leur traitement par trametinib. Les cas les plus sévères ont nécessité une hospitalisation, une interruption ou l'arrêt définitif du traitement. Les signes ou symptômes d'une rhabdomyolyse justifient une évaluation clinique appropriée et une prise en charge adaptée.
Pancréatite
Au cours des essais cliniques chez des patients adultes et pédiatriques, des cas de pancréatite ont été rapportés chez des patients traités par trametinib en association au dabrafenib (voir la rubrique Effets indésirables). Une douleur abdominale inexpliquée doit être rapidement investiguée par le dosage de l'amylase et la lipase sériques. Les patients doivent être étroitement surveillés lors de la reprise du traitement après un épisode de pancréatite.
Insuffisance rénale
Des insuffisances rénales ont été identifiées chez moins de 1 % des patients adultes traités par l'association trametinib et dabrafenib. Les cas observés chez les patients adultes étaient généralement associés à une pyrexie et une déshydratation et ont bien répondu à une interruption du traitement et aux mesures générales de prise en charge. Des néphrites granulomateuses ont également été rapportées chez des patients adultes. La créatinine sérique doit être régulièrement surveillée en cours de traitement. Si la créatinine augmente, une interruption de traitement pourra être nécessaire, selon la situation clinique. Le trametinib n'a pas été évalué chez des patients avec une insuffisance rénale (définie par une créatinine > 1,5 fois la valeur supérieure de la normale). La prudence est recommandée dans cette situation (voir la rubrique Propriétés pharmacocinétiques).
Evénements hépatiques
Des effets indésirables hépatiques ont été rapportés dans les essais cliniques réalisés chez les patients adultes et pédiatriques avec le trametinib en association au dabrafenib (voir la rubrique Effets indésirables). Il est recommandé de surveiller la fonction hépatique toutes les 4 semaines pendant les 6 premiers mois de traitement. La surveillance hépatique peut être poursuivie par la suite, selon la situation clinique.
Insuffisance hépatique
Le métabolisme hépatique et la sécrétion biliaire constituant les principales voies d'élimination du trametinib, le trametinib doit être administré avec prudence chez les patients présentant une insuffisance hépatique modérée à sévère (voir les rubriques Posologie et mode d'administration et Propriétés pharmacocinétiques).
Thrombose veineuse profonde (TVP)/ Embolie pulmonaire (EP)
Une embolie pulmonaire ou une thrombose veineuse profonde peut survenir. Si les patients développent des symptômes d'embolie pulmonaire ou de thrombose veineuse profonde tels qu'un essoufflement, une douleur thoracique ou un gonflement des bras ou des jambes, ils doivent consulter immédiatement un médecin. Le traitement doit être arrêté définitivement en cas d'embolie pulmonaire mettant en jeu le pronostic vital.
Affections gastro intestinales
Des cas de colites et d'entérocolites ont été rapportés chez les patients pédiatriques traités par le trametinib en association au dabrafenib (voir la rubrique Effets indésirables). Des colites et perforations gastro intestinales, parfois d'issue fatale, ont été rapportées chez des patients adultes. Le trametinib doit être administré avec précaution chez les patients présentant des facteurs de risque de perforation gastro intestinale, notamment des antécédents de diverticulite, des métastases du tube digestif et en cas de prise concomitante de médicaments présentant un risque connu de perforation gastro intestinale.
Sarcoïdose
Des cas de sarcoïdoses ont été signalés chez des patients adultes traités par trametinib en association au dabrafenib, impliquant principalement la peau, les poumons, les yeux et les ganglions lymphatiques. Dans la majorité des cas, le traitement par trametinib et dabrafenib a été maintenu. En cas de diagnostic de sarcoïdose, il convient d'envisager un traitement approprié.
Femmes en âge de procréer/ Fertilité chez les hommes
Avant d'initier le traitement chez des femmes en âge de procréer, une information appropriée sur les méthodes de contraception efficaces doit être donnée. Les femmes en âge de procréer recevant Trametinib 0,05 mg/mL doivent utiliser une méthode de contraception efficace tout au long de leur traitement et durant les 16 semaines suivant l'arrêt du traitement. Les patients de sexe masculin traités par trametinib en association au dabrafenib doivent être informés du risque potentiel d'altération de la spermatogénèse, qui peut être irréversible (voir la rubrique Fertilité, grossesse et allaitement).
Lymphohistiocytose hémophagocytaire
Après la mise sur le marché, une lymphohistiocytose hémophagocytaire (LHH) a été observée chez des patients traités par trametinib en association au dabrafenib. Il convient de faire preuve de prudence lorsque le trametinib est administré en association au dabrafenib. En cas de confirmation de LHH, l'administration de trametinib et de dabrafenib doit être interrompue, et un traitement de la LHH doit être instauré.
Excipients
Sulfobutyle bétadex de sodium
Trametinib 0,05 mg/mL poudre pour solution buvable contient de la cyclodextrine sulfobutyle bétadex de sodium (100 mg/mL.). Les cyclodextrines sont des excipients qui peuvent influencer les propriétés de la substance active et d'autres médicaments. Dans des études pré-cliniques chez l'animal, il y a eu des observations de toxicité rénale et d'ototoxicité chez les animaux ayant reçu des cyclodextrines par voie intraveineuse. Les aspects de sécurité des cyclodextrines ont été pris en compte lors du développement et de l'évaluation de la sécurité du médicament. Les données concernant la sécurité du médicament chez les enfants âgés de moins de 2 ans sont limitées.
Parahydroxybenzoate de méthyle
Ce médicament contient du parahydroxybenzoate de méthyle qui peut provoquer des réactions allergiques (éventuellement retardées).
Sodium
Ce médicament contient 1,98 mg de sodium par mL de solution buvable de Trametinib 0,05 mg/mL. Cela équivaut à 4 % de l'apport alimentaire quotidien maximal recommandé pour un adulte à la dose maximale quotidienne de 2 mg (40 mL) de trametinib.
Potassium
Ce médicament contient moins de 1 mmol de potassium (39 mg) par dose maximale quotidienne, c'est-à-dire qu'il est essentiellement « sans potassium ».
Résumé du profil de sécurité
Dans les essais cliniques pédiatriques avec le trametinib en association au dabrafenib, les effets indésirables les plus fréquents (rapportés à une fréquence ≥ 20 %) ont été : la pyrexie (65 %), l'éruption cutanée (47 %), la céphalée (39 %), les vomissements (38 %), la sécheresse de la peau (34 %), la fatigue (33 %), la diarrhée (30 %), les hémorragies (29 %), la neutropénie (25 %), la nausée (25 %), la dermatite acnéiforme (25 %), les douleurs abdominales (23 %) et la toux (21 %).
Les effets indésirables graves (grade 3/4) les plus fréquemment rapportés étaient : la neutropénie (15 %), la pyrexie (9 %), l'élévation des transaminases (6 %) et la prise de poids (4 %).
Le profil de sécurité chez les patients pédiatriques était en grande partie cohérent avec le profil de sécurité précédemment établi chez les patients adultes. Les effets indésirables supplémentaires suivants n'ont été rapportés jusqu'à présent que chez des patients adultes traités par trametinib en comprimés et dabrafenib en gélules : carcinome épidermoïde cutané, kératose séborrhéique, lymphoedème, sécheresse buccale, kératose actinique, hyperhydrose, photosensibilité (fréquent), mélanome, acrochordon, sarcoïdose, choriorétinopathie, pneumonie, insuffisance rénale, néphrite (peu fréquent), perforation gastro-intestinale (rare), lymphohistiocytose hémophagocytaire (rare), myocardite, syndrome de Stevens-Johnson, réaction médicamenteuse avec éosinophilie et symptômes systémiques (indéterminé).
Liste tabulée des effets indésirables
La sécurité de l'association de trametinib au dabrafenib a été établie à partir des données provenant de 171 patients pédiatriques pour l'analyse de sécurité poolée, comprenant deux études menées chez des patients atteints de tumeurs solides avancées présentant une mutation BRAF V600. Quatre patients (2,3 %) étaient âgés de 1 à < 2 ans, 39 patients (22,8 %) étaient âgés de 2 à < 6 ans,54 patients (31,6 %) étaient âgés de 6 à < 12 ans et 74 patients (43,3 %) étaient âgés de 12 à < 18 ans au moment du recrutement. La durée moyenne du traitement était de 80 semaines.
Les effets indésirables dans la population pédiatrique pour l'analyse de sécurité poolée sont listés ci- dessous (tableau 5) par classe de système d'organes MedDRA classées par fréquence et utilisant la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés en ordre décroissant de gravité.
Tableau 5 Effets indésirables rapportés dans la population pédiatrique pour l'analyse de sécurité du trametinib en association au dabrafenib (n= 171)
| Infections et infestations | |
| Très fréquent | Paronychie |
| Fréquent | Infection urinaire, cellulite, rhinopharyngite*1 |
| Tumeurs bénignes, malignes et non précisées (incluant les kystes et polypes) | |
| Fréquent | Papillome cutané |
| Affections hématologiques et du système lymphatique | |
| Très fréquent | Neutropenie*2, anémie, leucopénie* |
| Fréquent | Thrombocytopénie* |
| Affections du système immunitaire | |
| Fréquent | Hypersensibilité |
| Troubles du métabolisme et de la nutrition | |
| Fréquent | Déshydratation, diminution de l'appétit |
| Affections du système nerveux | |
| Très fréquent | Céphalée, sensations vertigineuses*3 |
| Affections oculaires | |
| Fréquent | Vision floue, trouble de l'acuité visuelle, uvéite*4 |
| Peu fréquent | Décollement de la rétine, oedème périorbitaire |
| Affections cardiaques | |
| Fréquent | Fraction d'éjection diminuée, bradycardie* |
| Affections vasculaires | |
| Très fréquent | Hémorragie*5 |
| Fréquent | Hypertension, hypotension |
| Affections respiratoires, thoraciques et médiastinales | |
| Très fréquent | Toux* |
| Fréquent | Dyspnée |
| Affections gastro-intestinales | |
| Très fréquent | Douleur abdominale*, constipation, diarrhée, nausée, vomissement |
| Fréquent | Pancréatite, stomatite |
| Peu fréquent | Colite* |
| Affections de la peau et du tissu sous-cutané | |
| Très fréquent | Dermatite acnéiforme *6, Sécheresse cutanée *7, prurit, éruption cutanée*8 |
| Fréquent | Dermatite exfoliative généralisée*9, alopécie, syndrome d'érythrodysesthésie palmo- plantaire, éruption pustulaire, folliculite, lésion cutanée, panniculite, érythème,hyperkératose |
| Peu fréquent | Fissures cutanées, sueurs nocturnes |
| Affections musculo-squelettiques et systémiques | |
| Très fréquent | Arthralgie, douleur des extrémités |
| Fréquent | Myalgie*, spasmes musculaires*10 |
| Troubles généraux et anomalies au site d'administration | |
| Très fréquent | Pyrexie*, fatigue*11, prise de poids |
| Fréquent | Inflammation des muqueuses, oedème du visage*, frissons, asthénie, oedème périphérique, syndrome pseudo-grippal |
| Investigations | |
| Très fréquent | Augmentation des transaminases*12 |
| Fréquent | Hyponatrémie, hypophosphatémie, hyperglycémie, augmentation des phosphatasesalcalines sanguines, augmentation des gamma GT, augmentation de la créatine phosphokinase sérique (CPK) |
| *Désigne un groupe de deux termes préférés MedDRA ou plus qui étaient considérés comme cliniquement similaires. 1 la rhinopharyngite comprend également la pharyngite 2 la neutropénie comprend également la diminution du nombre de neutrophiles et la neutropénie fébrile 3 les sensations vertigineuses comprennent également le vertige 4 l'uvéite comprend également l'iridocyclite 5 l'hémorragie comprend également l'épistaxis, l'hématurie, la contusion, l'hématome, l'augmentation du rapport international normalisé (INR), l'hémorragie anale, l'hémorragie au niveau du cathéter, l'hémorragie cérébrale, l'ecchymose, l'hématome extradurale, l'hémorragie gastro-intestinale, l'hématochézie (rectorragie), les pétéchies, l'hémorragie post-procédure, l'hémorragie rectale, la diminution du nombre de globules rouges, l'hémorragie gastro- intestinale haute et l'hémorragie utérin 6 la dermatite acnéiforme comprend également l'acné et l'acné pustuleuse. 7 la sécheresse cutanée comprend également la xérose et la xérodermie 8 l'éruption cutanée comprend également l'éruption maculo-papulaire, l'éruption pustuleuse, l'éruption érythémateuse, l'éruption papuleuse, l'éruption maculaire 9 la dermatite exfoliative généralisée comprend également l'exfoliation de la peau et la dermatite exfoliative 10 les spasmes musculaires comprennent également la raideur musculo-squelettique 11 la fatigue comprend également le malaise et l'asthénie 12 l'augmentation des transaminases, y compris augmentation de l'aspartate aminotransférase (ASAT) et de l'alanine aminotransférase (ALAT) | |
Description de certains effets indésirables
Prise de poids
Une prise de poids a été rapportée uniquement dans la population pédiatrique. Elle a été rapportée comme un effet indésirable chez 15 % des patients pédiatriques dont 4,1 % étaient de grade 3, avec un arrêt de traitement chez 1,2 % des patients. Le délai médian de survenue de la prise de poids chez les patients pédiatriques recevant le trametinib en association au dabrafenib était de 3,1 mois. Une augmentation du poids par rapport à la valeur initiale de ≥ 2 catégories de percentiles de l'IMC (indice de masse corporelle) pour l'âge a été observée chez 29,8 % des patients.
Hémorragie
Des évènements hémorragiques ont été observés chez 29 % des patients pédiatriques. L'événement hémorragique le plus fréquent (épistaxis) a été rapporté chez 17 % des patients pédiatriques. La plupart des événements étaient de grade 1 ou 2 et 1,2 % des cas survenus étaient de grade 3. Le délai médian de survenue de l'événement hémorragique chez les patients pédiatriques recevant le trametinib en association au dabrafenib était de 1,9 mois. Des événements hémorragiques, pour certains majeurs et des hémorragies d'issue fatale, sont survenus chez les patients adultes traités par trametinib en association au dabrafenib.
Le risque hémorragique peut être majoré par l'utilisation concomitante de médicaments antiplaquettaires ou anticoagulants. En cas d'hémorragie, les patients doivent être traités en fonction du tableau clinique (voir la rubrique Mises en garde spéciales et précautions d'emploi).
Réduction de la fraction d'éjection du ventricule gauche (FEVG) / dysfonction ventriculaire gauche
Des diminutions de la FEVG ont été rapportées chez 5,3 % des patients pédiatriques, dont des événements de grade 3 survenus chez < 1 % des patients. Le délai médian de survenue de réduction de la FEVG était d'environ 1 mois. Dans les essais cliniques chez les patients adultes, le délai médian d'apparition d'une dysfonction ventriculaire gauche, d'une insuffisance cardiaque ou d'une diminution de la FEVG a été d'environ 2 à 5 mois. Les patients présentant une FEVG inférieure à la valeur de la limite inférieure de la normale de l'établissement n'ont pas été inclus dans les essais cliniques avec trametinib. Le trametinib en association au dabrafenib doit être utilisé avec prudence chez les patients présentant des pathologies susceptibles d'affecter la fonction ventriculaire gauche (voir lesrubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Pyrexie
La survenue d'une pyrexie a été rapportée au cours des essais cliniques avec le trametinib. Cependant, l'incidence et la sévérité de la pyrexie étaient majorées avec l'association. Une pyrexie a été rapportée chez 65 % des patients pédiatriques dont 8,8 % des cas étaient de grade 3. Veuillez vous référer aux rubriques Mises en garde spéciales et précautions d'emploi et Effets indésirables du RCP de Dabrafenib en comprimés dispersibles.
Evènements hépatiques
Des effets indésirables hépatiques ont été rapportés chez les patients adultes et pédiatriques au cours des essais cliniques avec trametinib en monothérapie et en association au dabrafenib. Dans la population de sécurité pédiatrique, une augmentation des ALAT et des ASAT étaient des événements fréquents, 12,3 % et 15,2 % respectivement (voir la rubrique Mises en garde spéciales et précautions d'emploi). Les effets indésirables hépatiques augmentation des ALAT et augmentation des ASAT ont été les événements hépatiques les plus fréquents dans la population adulte et la majorité était de Grade 1 ou 2. Pour le trametinib en monothérapie, plus de 90 % de ces évènements hépatiques sont survenus dans les 6 premiers mois de traitements. Les évènements hépatiques ont été détectés dans les essais cliniques par une surveillance biologique toutes les 4 semaines. Il est recommandé pour les patients recevant trametinib en monothérapie ou en association au dabrafenib, de réaliser une surveillance de la fonction hépatique toutes les 4 semaines pendant les 6 premiers mois. La surveillance hépatique peut être poursuivie au delà en cas d'indication clinique (voir la rubrique Mises en garde spéciales et précautions d'emploi).
Modifications de la pression artérielle
Une hypertension a été rapportée chez 2,3 % des patients pédiatriques, dont des grades 3 survenus chez 1,2 % des patients. Le délai médian d'apparition de l'hypertension chez les patients pédiatriques était de 6,3 mois.
Une hypotension a été rapportée chez 3,5 % des patients pédiatriques, dont 2,3 % étaient de grade ≥ 3. Le délai médian d'apparition d'une hypotension chez les patients pédiatriques était de 1,5 mois.
La pression artérielle doit être mesurée à l'instauration du traitement et surveillée pendant le traitement par trametinib, et prise en charge par un traitement standard du contrôle de l'hypertension artérielle, si nécessaire (voir la rubrique Mises en garde spéciales et précautions d'emploi).
Atteinte pulmonaire interstitielle/Pneumopathie
Les patients adultes traités par trametinib sont susceptibles de développer une atteinte pulmonaire interstitielle ou pneumopathie. Le traitement par trametinib doit être suspendu chez les patients pour lesquels une pneumopathie ou une atteinte pulmonaire interstitielle est suspectée, incluant les patients présentant de nouveaux signes ou symptômes pulmonaires ou une progression de signes ou symptômes préexistants tels qu'une toux, une dyspnée, une hypoxie, un épanchement pleural ou des infiltrats pulmonaires, dans l'attente des résultats des investigations cliniques. Le traitement par trametinib doit être définitivement arrêté chez les patients avec un diagnostic avéré de pneumopathie ou d'atteinte pulmonaire interstitielle associée au traitement (voir les rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Troubles visuels
Des réactions ophtalmologiques ont été rapportés chez des patients pédiatriques traités par trametinib en association au dabrafenib, incluant des uvéites (1,8 %) et des iridocyclites (1,2 %). Des uvéites de grade 3 sont survenues chez < 1 % des patients pédiatriques. Des troubles, associés à des perturbations visuelles, tels qu'un décollement de l'épithélium pigmentaire de la rétine ou une occlusion de la veine rétinienne ont également été observés chez des patients adultes. Des symptômes tels qu'une vision floue, une baisse de l'acuité visuelle et d'autres troubles visuels ont été rapportés dans les essais cliniques réalisés avec le trametinib (voir les rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Eruption cutanée
Une éruption cutanée a été observée chez environ 47 % des patients pédiatriques dans les études avec l'association du trametinib au dabrafenib dans la population totale des études poolées pour l'analyse de sécurité. Dans la majorité des cas, les éruptions étaient de grade 1 ou 2 et n'ont pas nécessité de réduction de posologie ou d'interruption du traitement (voir les rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Rhabdomyolyse
Des cas de rhabdomyolyse ont été rapportés chez des patients adultes traités par trametinib. Les signes ou symptômes d'une rhabdomyolyse justifient une évaluation clinique appropriée et une prise en charge adaptée (voir la rubrique Mises en garde spéciales et précautions d'emploi).
Pancréatite
Des cas de pancréatite ont été rapportés chez 1,2 % des patients pédiatriques, dont < 1 % était de grade 3. Dans les essais cliniques chez les patients adultes, un événement de pancréatite est survenue le premier jour de traitement par dabrafenib chez un patient atteint de mélanome métastatique et a récidivé suite à un rechallenge à une posologie réduite. Toute douleur abdominale inexpliquée doit être investiguée, en incluant un dosage de l'amylase et de la lipase sériques. Les patients doivent être étroitement surveillés lors de la reprise du traitement après un épisode de pancréatite (voir la rubrique Mises en garde spéciales et précautions d'emploi).
Effets sur la maturation (sexuelle et squelettique) et le développement
Il n'existe actuellement pas de données sur les effets du traitement par trametinib associé au dabrafenib sur la maturation (sexuelle et squelettique) et le développement des patients pédiatriques (voir rubrique Données de sécurité préclinique).
Insuffisance rénale
Une insuffisance rénale a été rapportée avec le dabrafenib en association au trametinib.
Une insuffisance rénale due à une azotémie extra-rénale associée à la
pyrexie ou à une néphrite granulomateuse s'est révélée peu fréquente
chez les patients adultes ; toutefois, le dabrafenib n'a pas été étudié
chez les patients ayant une insuffisance rénale (définie par une
créatinine > 1,5 fois la valeur supérieure de la normale). La
prudence est recommandée dans cette situation (voir la rubrique Mises en garde spéciales et précautions d'emploi).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé doivent déclarer tout effet indésirable suspecté selon les modalités définies dans le Protocole d'utilisation thérapeutique et de recueil de données (voir PUT RD).
AVANT la mise en route du
traitement : confirmer par un test validé que le patient est bien atteint d'une
tumeur avec la mutation BRAF V600E.
SURVEILLANCE DU TRAITEMENT :
-
Cardiaque : la FEVG doit être évaluée avant l'instauration du traitement, un
mois après, puis environ tous les 3 mois durant le traitement.
- Tensionnelle
: la pression artérielle doit être mesurée à l'instauration du traitement et
pendant le traitement.
- Créatinine sérique.
- Hépatique : la fonction
hépatique doit être surveillée toutes les 4 semaines pendant les 6 premiers mois
de traitement puis selon la situation clinique.
- Cutanée avant l'initiation
du traitement, puis mensuellement pendant le traitement et jusqu'à 6 mois après
le traitement.
De nouveaux mélanomes primitifs ont été rapportés chez les
patients recevant trametinib en association au dabrafenib.
Informer les
femmes en âge de procréer d' UTILISER UNE METHODE DE CONTRACEPTION NON HORMONALE
EFFICACE pendant le traitement et durant les 4 mois suivant son arrêt
(l'utilisation en association au dabrafenib peut diminuer l'efficacité de la
contraception hormonale, une méthode alternative de contraception, telle qu'une
méthode de type barrière, doit être utilisée pendant le traitement par
l'association trametinib/dabrafenib).
Informer les patients de sexe masculin
traités par le trametinib en association au dabrafenib du risque potentiel
d'altération de la spermatogénèse, qui peut être irréversible.
INFORMER LES PATIENTS :
- des signes et symptômes des réactions cutanées;
- du risque potentiel de fatigue, de sensations vertigineuses ou de problèmes oculaires affectant l'aptitude à conduire des véhicules et à utiliser des machines.
Femmes en âge de procréer - Contraception chez les femmes
Les femmes en âge de procréer doivent utiliser une méthode de contraception efficace tout au long de leur traitement par trametinib, et durant les 16 semaines suivant l'arrêt du traitement.
L'utilisation en association au dabrafenib peut diminuer l'efficacité de la contraception hormonale, une méthode alternative de contraception, telle qu'une méthode de type barrière, doit être utilisée pendant le traitement par l'association trametinib/dabrafenib. Veuillez-vous référer au RCP de Dabrafenib 10 mg comprimé dispersible pour plus d'informations.
Grossesse
Il n'y a pas de données sur l'utilisation du trametinib chez la femme enceinte. Les études réalisées chez l'animal ont montré une toxicité sur la reproduction (voir la rubrique Données de sécurité préclinique). Le trametinib ne doit pas être administré aux femmes enceintes sauf si le bénéfice potentiel pour la mère l'emporte sur le risque éventuel pour le foetus. Si le trametinib est utilisé pendant la grossesse ou si une grossesse survient pendant le traitement par trametinib, la patiente devra être informée du risque potentiel pour le foetus.
Allaitement
Le passage du trametinib dans le lait maternel n'est pas connu. Un risque pour l'enfant allaité ne peut être exclu. Le trametinib ne doit pas être administré chez les mères allaitant. La décision d'interrompre soit l'allaitement soit le traitement par le trametinib devra prendre en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la mère.
Fertilité
Aucune donnée n'est disponible chez l'être humain avec le trametinib. Chez l'animal, aucune étude sur la fertilité n'a été réalisée mais des effets indésirables ont été observés au niveau des organes reproducteurs féminins (voir la rubrique Données de sécurité préclinique). Le trametinib peut avoir un effet délétère sur la fertilité chez les humains.
Pour les hommes traités par trametinib en association au dabrafenib
Des effets sur la spermatogénèse ont été observés chez les animaux traités par le dabrafenib. Les patients de sexe masculin traités par le trametinib en association au dabrafenib doivent être informés du risque potentiel d'altération de la spermatogénèse, qui peut être irréversible. Pour plus d'informations, veuillez-vous référer au RCP de Dabrafenib 10 mg comprimé dispersible.
Les études d'interaction n'ont été réalisées que chez l'adulte.
Effets d'autres médicaments sur le trametinib
Le trametinib étant principalement métabolisé par une désacétylation induite par des enzymes hydrolytiques (par exemple les carboxylestérases), ses caractéristiques pharmacocinétiques sont peu susceptibles d'être modifiées par d'autres agents via des interactions métaboliques (voir rubrique Propriétés pharmacocinétiques). Des interactions entre médicaments médiées par ces enzymes hydrolytiques ne peuvent être exclues et pourraient avoir un impact sur l'exposition au trametinib.
Trametinib est un substrat du transporteur d'efflux P-gp in vitro. Comme il ne peut être exclu qu'une forte inhibition de la P-gp hépatique puisse résulter en une augmentation de l'exposition au trametinib, une attention particulière est recommandée lorsque trametinib est administré en association à d'autres médicaments qui sont de puissants inhibiteurs de la P-gp (par exemple vérapamil, cyclosporine, ritonavir, quinidine, itraconazole).
Effets du trametinib sur d'autres médicaments
Sur la base des données in vitro et in vivo, le trametinib est peu susceptible de modifier significativement les caractéristiques pharmacocinétiques d'autres médicaments via les enzymes ou les transporteurs du CYP (voir la rubrique Propriétés pharmacocinétiques). Le trametinib peut entraîner une inhibition transitoire des substrats du BCRP (par exemple : pitavastatine) au niveau intestinal, qui peut toutefois être minimisée par une répartition appropriée des prises (respect d'un intervalle de 2 heures) de ces médicaments et du trametinib.
Sur la base de données cliniques, aucune perte d'efficacité des contraceptifs hormonaux n'est attendue lorsqu'ils sont administrés concomitamment au trametinib(voir la rubrique Propriétés pharmacocinétiques). Cependant, l'utilisation avec le dabrafenib peut rendre les contraceptifs hormonaux moins efficaces.
Veuillez vous reporter également aux recommandations sur les interactions médicamenteuses pour le dabrafenib figurant dans les rubriques Mises en garde spéciales et précautions d'emploi et Interactions avec d'autres médicaments et autres formes d'interactions du RCP de dabrafenib en comprimés dispersibles.
Effets de l'excipient sulfobutyle bétadex de sodium sur d'autres médicaments avec une biodisponibilité faible et un indice thérapeutique étroit : la solution buvable de trametinib contient 100 mg/mL de sulfobutyle bétadex de sodium qui peut potentiellement affecter la solubilité et la biodisponibilité de médicaments administrés concomitamment par voie orale. La solution buvable de trametinib est à administrer avec prudence avec d'autres médicaments ayant une faible biodisponibilité et un indice thérapeutique étroit.
Le traitement par Trametinib 0,05 mg/mL doit être initié et supervisé par un médecin expérimenté dans l'utilisation des traitements anticancéreux.
Avant de prendre Trametinib 0,05 mg/mL les patients doivent avoir la confirmation par un test validé, que leur tumeur est porteuse de la mutation BRAF V600E.
Trametinib 0,05 mg/mL est utilisé en association au dabrafenib en comprimés dispersibles. Veuillez vous référer au Résumé des Caractéristiques du Produit (RCP) pour la posologie de dabrafenib en comprimés dispersibles.
Posologie
La dose recommandée de Trametinib 0,05 mg/mL en une prise par jour est déterminée par l'âge et le poids (Tableau 1).
Tableau 1 Schéma posologique en fonction de l'âge et du poids
| | Age < 6 ans | Age ≥ 6 ans |
| Dose recommandée | 0,032 mg/kg/jour en une prise par jour | 0,025 mg/kg/jour en une prise par jour |
La dose recommandée pour les patients pesant moins de 8 kg n'a pas été établie.
Veuillez vous référer aux rubriques « Posologie » et « Mode d'administration » du RCP du dabrafenib en comprimés dispersibles pour les instructions sur la prise du traitement par dabrafenib lorsqu'il est utilisé en association avec Trametinib poudre pour solution buvable.
La dose journalière de trametinib ne doit pas dépasser 2 mg.
Chaque mL de la solution reconstituée contient 0,05 mg de trametinib.
Durée du traitement
Le traitement par Trametinib 0,05 mg/mL doit être poursuivi jusqu'à progression de la maladie ou la survenue d'une toxicité inacceptable. Il existe des données limitées chez les patients âgés de plus de 18 ans atteints de gliome, par conséquent la poursuite du traitement à l'âge adulte devra être décidée en fonction du rapport bénéfice/risque individuel évalué par le médecin.
Omission ou retard d'administration d'une dose
Si une dose de Trametinib 0,05 mg/mL est oubliée, elle ne doit être prise que s'il reste plus de 12 heures avant la prise suivante prévue. Si le patient vomit après avoir pris une dose de Trametinib 0,05 mg/mL, une dose supplémentaire ne doit pas être prise et la prochaine administration doit avoir lieu à la prochaine prise prévue.
Adaptations posologiques
La prise en charge des effets indésirables peut nécessiter une réduction de dose, une interruption temporaire du traitement ou un arrêt du traitement (voir tableaux 2 et 3).
Si des toxicités liées au traitement surviennent, alors les posologies de dabrafenib et de trametinib doivent simultanément être réduites, interrompues ou arrêtées. Les exceptions pour lesquelles des adaptations posologiques sont nécessaires pour un seul des deux traitements sont détaillées ci-dessous pour l'uvéite, les tumeurs malignes non cutanées ayant une mutation RAS positive (toxicités principalement liées au dabrafenib), la réduction de la fraction d'éjection du ventricule gauche (FEVG), l'occlusion de la veine rétinienne (OVR), le décollement de l'épithélium pigmentaire de la rétine (DEP), et maladie pulmonaire interstitielle/pneumopathie (toxicités principalement liées au trametinib).
Il n'est pas recommandé d'effectuer des adaptations posologiques ou d'interrompre le traitement en cas de survenue de tumeurs cutanées malignes (voir le RCP de Dabrafenib 10 mg comprimés dispersibles).
Tableau 2 Schéma d'adaptation posologique selon le grade des effets indésirables (excepté la pyrexie)
| Grade (CTCAE)* | Recommandations de modifications de la posologie du dabrafenib |
| Grade 1 ou Grade 2 (Tolérable) | Maintien de la dose et surveillance clinique appropriée. |
| Grade 2 (Intolérable)ou Grade 3 | Interruption du traitement jusqu'au retour à la normale ou à une toxicité de Grade 1 et reprise du traitement à une dose réduite d'un palier Se référer au Tableau 3 pour les adaptations posologiques |
| Grade 4 | Arrêt définitif ou interruption du traitement jusqu'au retour à la normale ou à un Grade 1 et reprise du traitement à une dose réduite d'un palier Se référer au Tableau 3 pour les adaptations posologiques |
| *L'intensité des effets indésirables cliniques est cotée selon les critères communs de terminologie pour les événements indésirables (Common Terminology Criteria for Adverse Events ; CTCAE). | |
Les recommandations de réductions de doses à environ 75% de la dose recommandée (1er niveau de réduction de dose) et à environ 50% de la dose recommandée (2ème niveau de réduction de dose) sont présentées dans le Tableau 3.
Tableau 3 Recommandations de réduction de dose en cas d'effets indésirables
| | Age < 6 ans | Age ≥ 6 ans |
| Dose initiale | 0,032 mg/kg/jour en une prise par jour | 0,025 mg/kg/jour en une prise par jour |
| 1er palier de réduction de dose | 0,025 mg/kg/jour en une prise par jour | 0,017 mg/kg/jour en une prise par jour |
| 2ème palier de réduction de dose | 0,016 mg/kg/jour en une prise par jour | 0,013 mg/kg/jour en une prise par jour |
| Un ajustement posologique de Trametinib 0,05 mg/mL inférieur à 50% de la dose recommandée n'est pas recommandé. | ||
Lorsque les effets indésirables d'un patient sont bien pris en charge, une ré-augmentation de dose peut être envisagée, en respectant les paliers utilisés au moment de la réduction de la dose. La posologie de trametinib ne doit pas excéder la dose recommandée dans le Tableau 1.
Modification de la posologie en cas d'effets indésirables particuliers
Pyrexie
Si la température corporelle du patient est ≥ 38oC, le traitement doit être interrompu. En cas de récurrence, le traitement peut également être interrompu au premier symptôme de pyrexie. Un traitement par antipyrétiques tels que l'ibuprofène ou le paracétamol doit être instauré. L'utilisation de corticostéroïdes par voie orale doit être envisagée dans le cas où les antipyrétiques s'avèrent insuffisants. Des signes et symptômes d'infection doivent être recherchés et si nécessaire, les patients doivent être traités conformément aux pratiques locales (voir la rubrique Mises en garde spéciales et précautions d'emploi). Le traitement est à reprendre si le patient n'a pas eu de symptôme pendant au moins 24 heures, soit (1) à la même dose, soit (2) au palier de dose inférieur, si la fièvre est récurrente et/ou était accompagnée d'autres symptômes sévères dont une déshydratation, une hypotension ou une insuffisance rénale.
Exceptions concernant les modifications de posologie (lorsque la réduction de dose s'appliqueuniquement pour un des deux traitements) spécifiques à certains effets indésirables
Diminution de la fraction d'éjection du ventricule gauche (FEVG)/ Dysfonction ventriculaire gauche
Le traitement par trametinib doit être interrompu chez les patients
présentant une diminution absolue > 10 % et asymptomatique de la
fraction d'éjection du ventricule gauche par rapport à la valeur
initiale et avec une valeur de la fraction d'éjection inférieure à la
limite inférieure de la normale de l'établissement (voir la rubrique Mises en garde spéciales et précautions d'emploi).
Aucune adaptation posologique de dabrafenib n'est requise. Si la
fraction d'éjection ventriculaire gauche se rétablit, le traitement par
trametinib peut être repris mais à une dose réduite d'un palier et sous
surveillance clinique étroite (voir la rubrique Mises en garde spéciales et précautions d'emploi).
Le traitement par le trametinib doit être définitivement arrêté chez les patients qui présentent une dysfonction ventriculaire gauche de Grade 3 ou 4 ou une réduction cliniquement significative de la fraction d'éjection ventriculaire gauche qui ne s'est pas rétablie dans un délai de 4 semaines (voir la rubrique Mises en garde spéciales et précautions d'emploi).
Occlusion de la veine rétinienne et décollement de l'épithélium pigmentaire de la rétine
Si les patients signalent l'apparition d'un trouble de la vision, comme
une diminution de la vision centrale, une vision floue ou une perte de
l'acuité visuelle durant le traitement par trametinib, une évaluation
ophtalmologique doit être rapidement réalisée. Le traitement par
trametinib doit être arrêté définitivement si une occlusion de la veine
rétinienne est diagnostiquée durant le traitement. Aucune adaptation
posologique de dabrafenib n'est requise. Si un décollement de
l'épithélium pigmentaire de la rétine (décollement de la rétine) est
diagnostiqué durant le traitement, suivre les modifications de
posologie du trametinib indiquées dans le Tableau 4 (voir la rubrique Mises en garde spéciales et précautions d'emploi).
Tableau 4 Recommandations d'adaptation posologique du trametinib en cas de décollement de la rétine
| Décollement de la rétine Grade 1 | · Poursuivre le traitement avec une évaluation mensuelle de la rétine jusqu'à résolution du décollement. Si le décollement de la rétine s'aggrave, suivre les instructions ci-dessous et suspendre le traitement par trametinib jusqu'à 3 semaines. |
| Décollement de la rétine Grade 2-3 | · Suspendre le traitement par trametinib jusqu'à 3 semaines. |
| Décollement de la rétine Grade 2-3 revenant à la normale ou Grade 1 en 3 semaines | · Reprendre le traitement par trametinib à une posologie plus faible (voir Tableau 3) ou arrêter le traitement par trametinib chez les patients recevant la posologie la plus faible. |
| Décollement de la rétine Grade 2-3 sans retour à la normale ou Grade 1 en 3 semaines | · Arrêt définitif du traitement par trametinib. |
Atteinte pulmonaire interstitielle/pneumopathie
Le traitement par trametinib doit être suspendu chez les patients pour
lesquels une atteinte pulmonaire interstitielle ou pneumopathie est
suspectée, incluant les patients présentant de nouveaux symptômes ou
signes pulmonaires ou une progression de symptômes ou signes
préexistants incluant une toux, une dyspnée, une hypoxie, un
épanchement pleural ou des infiltrats pulmonaires, dans l'attente des
résultats des investigations cliniques. Le traitement par trametinib
doit être définitivement arrêté chez les patients avec un diagnostic
avéré de pneumopathie ou atteinte pulmonaire interstitielle associée au
traitement. Aucune adaptation de posologie de dabrafenib n'est requise.
Uvéite
Aucune adaptation de la posologie n'est requise en cas d'uvéite tant
que des traitements locaux efficaces peuvent contrôler l'inflammation
oculaire. Si l'uvéite ne répond pas aux traitements oculaires locaux,
dabrafenib doit être interrompu jusqu'à la résolution de l'inflammation
oculaire puis dabrafenib doit être réintroduit à une dose réduite d'un
palier. Aucune modification de dose de trametinib n'est requise (voir
la rubrique Mises en garde spéciales et précautions d'emploi).
Tumeurs malignes non cutanées RAS mutées
Les bénéfices et les risques doivent être envisagés avant de
continuer le traitement par dabrafenib chez les patients présentant une
tumeur maligne non cutanée associée à une mutation RAS. Aucune
modification de dose de trametinib n'est requise voir la rubrique Mises en garde spéciales et précautions d'emploi).
Populations particulières
Insuffisance hépatique
Aucune adaptation posologique n'est nécessaire chez les patients ayant
une insuffisance hépatique légère. Les données disponibles issues d'une
étude clinique de pharmacologie indiquent un impact limité de
l'insuffisance hépatique modérée à sévère sur l'exposition au
trametinib (voir la rubrique Propriétés pharmacocinétiques). Le trametinib doit être utilisé avec prudence en cas d'insuffisance hépatique modérée à sévère.
Insuffisance rénale
Aucune adaptation posologique n'est nécessaire chez les patients ayant
une insuffisance rénale légère à modérée. L'absence de données
concernant le trametinib chez les patients ayant une insuffisance
rénale sévère ne permet pas d'établir de recommandations d'adaptation
de posologie (voir la rubrique Propriétés pharmacocinétiques). Le trametinib doit être utilisé avec prudence chez les patients présentant une insuffisance rénale sévère.
Population pédiatrique
La sécurité et l'efficacité de l'association du trametinib au
dabrafenib chez les enfants âgés de moins de 1 an n'ont pas été
établies. Aucune donnée n'est disponible. Des études réalisées chez de
jeunes animaux ont mis en évidence des effets indésirables du
dabrafenib qui n'ont pas été observés chez les animaux adultes (voir la
rubrique Données de sécurité préclinique).Les données de sécurité à long-terme chez les patients pédiatriques sont actuellement limitées.
Mode d'administration
Voie orale.
Il est recommandé que la reconstitution de la solution buvable soit effectuée par un professionnel de santé (par exemple un pharmacien) avant d'être délivrée au patient. Il est recommandé que le professionnel de santé explique comment administrer la dose quotidienne prescrite de solution buvable avant la première administration.
Trametinib 0,05 mg/mL doit être administré sans nourriture, au moins une heure avant, ou deux heures après un repas. Du lait maternel et/ou infantile peut être donné à la demande si le patient ne peut pas rester à jeun. Une durée de jeune plus courte pourrait réduire la biodisponibilité du trametinib (voir la rubrique Propriétés pharmacocinétiques).
Il est recommandé de prendre la dose de trametinib approximativement à la même heure chaque jour, en utilisant une seringue orale réutilisable. La dose quotidienne de trametinib doit être prise tous les jours au même moment que la dose du matin ou du soir de dabrafenib en comprimés dispersibles.
Si le patient est incapable d'avaler et qu'il a une sonde nasogastrique in situ, la solution buvable de trametinib peut être administré via la sonde. La sonde doit être rincée à l'eau après l'administration de Trametinib 0,05 mg/mL.
Pour les instructions concernant la reconstitution du médicament avant administration, veuillez vous référer au « Guide de reconstitution à destination des personnes en charge du traitement Trametinib en solution buvable ».
Durée de conservation :
Poudre pour solution buvable 3 ans
Solution buvable reconstituée
Jeter la solution non utilisée dans les 35 jours après la reconstitution.
Précautions particulières de conservation :
Avant reconstitution : A conserver au réfrigérateur (entre 2°C et 8°C). Ne pas congeler.
Après reconstitution : A conserver à une température ne dépassant pas 25°C. Ne pas congeler.
Sans objet.
Il n'y a pas d'antidote spécifique en cas de surdosage. En cas de surdosage, le patient doit recevoir un traitement symptomatique approprié et une surveillance adéquate si nécessaire.
Classe pharmacothérapeutique : Agent antinéoplasique, inhibiteurs de protéine kinase, Inhibiteurs de MEK, Code ATC : L01EE01
Mécanisme d'action
Le trametinib est un inhibiteur allostérique, réversible et hautement sélectif de l'activation du signal régulé par MEK 1 (mitogen-activated extracellular signal regulated kinase 1) et MEK2 et de l'activité des kinases. Les protéines MEK sont des composants de la voie régulée par la kinase ERK (extracellular signal related kinase). Dans les cancers chez l'Homme, cette voie est souvent activée par des formes mutées de BRAF qui activent MEK. Le trametinib inhibe l'activation de MEK par BRAF et inhibe l'activité de la kinase MEK.
Dabrafenib est un inhibiteur des RAF kinases. Les mutations oncogéniques BRAF conduisent à une activation constitutive de la voie de signalisation RAS/RAF/MEK/ERK. Ainsi, trametinib et dabrafenib inhibent deux kinases de cette voie, MEK et RAF, conduisant ainsi à l'inhibition concomitante par l'association de la voie de signalisation. L'association de trametinib au dabrafenib a montré une activité anti-tumorale in vitro sur les lignées cellulaires de cancer BRAF V600 mutées et retarde l'apparition de résistance in vivo des xénogreffes BRAF V600 mutées.
Efficacité et sécurité clinique
Population pédiatrique
L'efficacité clinique et la sécurité du traitement par dabrafenib en association au trametinib chez les patients pédiatriques âgés de 1 à < 18 ans atteints d'un gliome porteur de la mutation BRAF V600 ont été évaluées dans le cadre de l'essai clinique de phase II (EudraCT 2015-004015-20), en ouvert, multicentrique. Les patients atteints de gliome de bas grade (grades 1 et 2 de l'OMS 2016) qui nécessitaient un traitement par voie systémique ont été randomisés selon un rapport 2:1 pour recevoir du dabrafenib avec du trametinib ou du carboplatine avec de la vincristine, et des patients atteints de gliome de haut grade en rechute ou réfractaire (grades 3 et 4 de l'OMS 2016) ont été recrutés dans un bras unique avec dabrafenib plus trametinib.
Le statut de mutation BRAF a été identifié de manière prospective via un test local ou un test de réaction en chaîne par polymérase (PCR) en laboratoire centralisé lorsqu'un test local n'était pas possible. De plus, un test rétrospectif des échantillons de tumeurs disponibles a été effectué en laboratoire centralisé pour confirmer la mutation BRAF V600E.
La posologie du dabrafenib et du trametinib dans l'essai clinique dépendait de l'âge et du poids, le dabrafenib étant administré par voie orale à 2,625 mg/kg deux fois par jour pour les enfants de moins de 12 ans et à 2,25 mg/kg deux fois par jour pour les enfants de 12 ans et plus ; le trametinib a été administré par voie orale à 0,032 mg/kg une fois par jour pour les enfants de moins de 6 ans et à 0,025 mg/kg une fois par jour pour les enfants de 6 ans et plus. Les doses de dabrafenib ont été limitées à 150 mg deux fois par jour et les doses de trametinib à 2 mg une fois par jour. La posologie du carboplatine et la vincristine dépendait de l'âge et de la surface corporelle avec des doses de 175 mg/m² et 1,5 mg/m², respectivement, en perfusions hebdomadaires. Le carboplatine et la vincristine ont été administrés en cure d'induction de 10 semaines suivi de huit cures d'entretien de 6 semaines.
Le critère principal d'évaluation de l'efficacité dans les deux cohortes était le taux de réponse global (TRG, somme des réponses complètes/RC et partielles/RP confirmées) par une évaluation indépendante basé sur les critères RANO (2017) pour la cohorte GBG, et RANO (2010) pour la cohorte GHG. L'analyse primaire a été réalisée lorsque tous les patients dans les deux cohortes avaient terminé au moins 32 semaines de traitement.
Gliome de bas grade pédiatrique (Grades 1 et 2 de l'OMS) porteur de la mutation BRAF
Dans la cohorte des gliomes de bas grade, 110 patients ont été randomisés pour recevoir dabrafenib avec le trametinib (n=73) ou carboplatine plus vincristine (n=37). L'âge médian était de 9,5 ans, avec 34 patients (30,9 %) âgés de 12 mois à < 6 ans, 36 patients (32,7 %) âgés de 6 à < 12 ans et 40 patients (36,4 %) âgés de 12 à < 18 ans ; 60 % étaient de sexe féminin. La majorité des patients (80 %) avait un gliome de grade 1 au diagnostic initial. Les pathologies les plus fréquentes étaient l'astrocytome pilocystique (30,9 %), le gangliome (27,3 %) et le GBG sans indication (18,2 %). Des sites métastatiques étaient présents chez 9 patients (8,2 %). Une chirurgie antérieure avait été rapportée chez 91 patients (82,7 %), dont une résection comme dernière procédure chirurgicale chez 28 patients (25,5 %). L'utilisation de corticostéroïdes par voie systémique a été rapporté chez 37 patients (33,6 %).
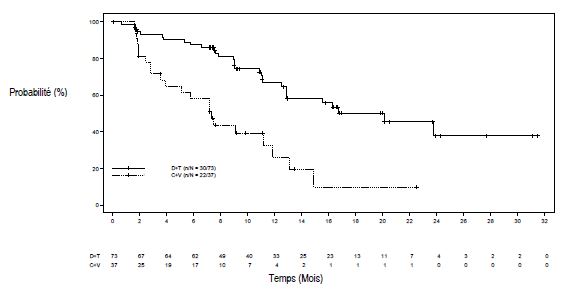
Le TRG dans le bras dabrafenib plus trametinib a montré une amélioration statistiquement significative par rapport au bras carboplatine plus vincristine (Tableau 6).
Lors de l'analyse principale, conduite après que tous les patients aient terminé 32 semaines de traitement ou aient arrêté le traitement prématurément, les données de survie globale (SG) étaient encore immatures (un décès a été rapporté dans le bras carboplatine plus vincristine (C+V)).
Tableau 5 Réponse et survie sans progression dans l'étude G2201 (cohorte GBG)
| | Dabrafenib + Trametinib (D+T)N=73 | Carboplatine + Vincristine (C+V)N=37 |
| Meilleure réponse globale | | |
| Réponse complète (RC), n (%) | 2 (2,7) | 1 (2,7) |
| Réponse partielle (RP), n (%) | 32 (43,8) | 3 (8,1) |
| Maladie stable (MS), n (%) | 30 (41,1) | 15 (40,5) |
| Progression de la maladie (PD), n (%) | 8 (11,0) | 12 (32,4) |
| Indéterminé, n (%) | 1 (1,4) | 6 (16,2)1 |
| Taux de réponse globale | | |
| TRG (RC+RP), IC à 95% | 46,6% (34,8 - 58,6%) | 10,8% (3,0 - 25,4%) |
| Odds ratio2 | 7,19 (2,3 - 22,4), p<0,001 | |
| Différence de risque | 35,8 % (20,6 - 51,0) | |
| Survie sans progression (SSP) | | |
| SSP (mois), (IC à 95 %) | 20,1 (12,8 - NE) | 7,4 (3,6 - 11,8) |
| Hazard ratio (IC à 95%), p-value | 0,31 (0,17 - 0,55), p<0,001 | |
| NE = non évaluable 1 4 patients randomisés dans le bras C+V sont sortis de l'essai avant de recevoir le traitement 2 Odds ratio (D+T vs C+V) et un IC à 95 % venant d'une régression logistique avec le traitement comme seule covariable, c'est-à-dire que c'est la chance d'observer une réponse dans le bras D+T comparé à la chanced'observer une réponse dans le bras C+V. L'odds ratio > 1 est en faveur de D+T. | ||
Figure 1 Courbe de Kaplan-Meier de la survie sans progression pour l'étude G2201 (cohorte GBG)
Gliome de haut grade pédiatrique (Grades 3 et 4 de l'OMS) porteur de la mutation BRAF
Dans la cohorte simple bras pour les gliomes de haut grade, 41 patients atteints de gliome de haut grade en rechute ou réfractaire ont été recrutés et traités par dabrafenib plus trametinib pendant une durée médiane de 72,7 semaines. L'âge médian était de 13,0 ans, avec 5 patients (12,2 %) âgés de 12 mois à < 6 ans, 10 patients (24,4 %) âgés de 6 à < 12 ans et 26 patients (63,4 %) âgés de 12 à < 18 ans ; 56 % étaient de sexe féminin. Le grade histologique au diagnostic initial était un grade 4 pour 20 patients (48,8 %), un grade 3 pour 13 patients (31,7 %), un grade 2 pour 4 patients (9,8 %), un grade 1 pour 3 patients (7,3 %) et le grade était manquant pour 1 patient (2,4 %). Les pathologies les plus fréquentes étaient le glioblastome multiforme (31,7 %), le xanthoastrocytome anaplasique pléomorphe (14,6 %), le GHG sans indication (9,8 %) et le xanthoastrocytome pléomorphe (9,8 %).
Une chirurgie antérieure avait été rapportée chez 40 patients (97,6 %), dont une résection comme dernière procédure chirurgicale chez 24 patients (58,5 %). Une chimiothérapie antinéoplasique antérieure a été rapportée chez 33 patients (80,5 %). Une radiothérapie antérieure a été rapportée chez 37 patients (90,2 %). L'utilisation de corticostéroïdes par voie systémique pendant le traitement a été rapportée chez 20 patients (48,8 %).
Le TRG dans cette cohorte était de 56,1 % (23/41), avec un IC à 95 % (39,7 - 71,5 %) : RC chez 12 patients et RP chez 11 patients (26,8 %). La durée de réponse médiane (DDR) était de 22,2 mois (IC à 95 % : 7,6 - NE), avec 15 patients (65,2 %) censurés au moment de l'analyse principale.
Les propriétés pharmacocinétiques du trametinib ont principalement été déterminées chez des patients adultes en utilisant la formulation solide (comprimé). La pharmacocinétique du trametinib après une administration unique ou répétée ajustée en fonction du poids a également été évaluée chez 244 patients pédiatriques. Les caractéristiques pharmacocinétiques (taux d'absorption du médicament et clairance du médicament) du trametinib chez les patients pédiatriques étaient comparables à celles des adultes. Il a été constaté que le poids influençait la clairance orale du trametinib, contrairement à l'âge. Les expositions pharmacocinétiques au trametinib à la posologie recommandée ajustée en fonction du poids chez les patients pédiatriques se situaient dans les limites de celles observées chez les adultes.
Absorption
La solution buvable de trametinib a été absorbée rapidement avec un temps médian pour atteindre la concentration plasmatique maximale (Tmax) de 1 heure après administration. La biodisponibilité absolue moyenne du trametinib comprimé était de 72%. Dans une étude de biodisponibilité relative comparant la formulation solution buvable et la formulation comprimé après une administration unique chez l'adulte à jeun, l'ASC(0-inf) , l'ASC(0-dernier) ,et la Cmax étaient augmentées de 12 %, 10 % et 71 % respectivement après l'administration de la solution buvable par rapport à la formulation comprimé.
Dans l'essai clinique pédiatrique pivot, la moyenne géométrique de la Cmax à l'état d'équilibre (%CV) et l'ASC(0-t) étaient de 22,7 ng/ml (41,1 %) et 339 ng*h/ml (22,2 %) dans la cohorte GBG et 21,3 ng/ml(36,3 %) et 307 ng*hr/ml (22,8 %) dans la cohorte de GHG.
Lors d'administrations répétées quotidiennes, le trametinib s'accumule. Un rapport moyen d'accumulation de 6,0 a été observé pour la formulation à 2 mg en comprimés une fois par jour. L'état d'équilibre a été atteint au jour 15.
Effet de la nourriture
L'administration d'une dose unique de trametinib (formulation en comprimés) avec un repas riche en graisses et en calories a réduit sa biodisponibilité (Cmax et ASC diminuées respectivement de 70 % et 10 %) par rapport à un état à jeun (voir rubriques Posologie et mode d'administration et Interactions avec d'autres médicaments et autres formes d'interactions). Bien que les données pour la formulation en solution buvable ne soient pas encore disponibles, il est attendu que l'effet de la nourriture sur la solution buvable reconstituée soit de même ampleur.
Le trametinib se lie à 97,4 % aux protéines plasmatiques humaines. Le volume de distribution du trametinib à l'état d'équilibre après administration d'une microdose intraveineuse de 5 µg est d'environ 1200 L.
Biotransformation
Les études in vitro et in vivo ont montré que le trametinib était principalement métabolisé par désacétylation seule ou en association avec une mono-oxygénation. Le métabolite désacétylé était ensuite métabolisé par glucuronidation. L'oxydation par CYP3A4 est considérée comme une voie mineure de métabolisation. La désacétylation est réalisée par les carboxylestérases 1b, 1c et 2, avec la participation potentielle d'autres enzymes hydrolytiques.
Suite à l'administration d'une dose unique et de doses répétées, le trametinib en tant que composé parent est le principal composé circulant dans le plasma.
Élimination
La demi vie terminale du trametinib après administration d'une dose unique est de 127 heures (5,3 jours). La clairance apparente du trametinib chez les patients adultes était 5,07 L /h (CV de 25 %).
Après l'administration par voie orale d'une dose unique de trametinib radiomarqué en solution, la quantité totale de la dose retrouvée était faible (< 50 %) d'après les prélèvements effectués sur une période de 10 jours, du fait d'une longue demi vie d'élimination. Les substances liées au trametinib étaient majoritairement excrétées dans les fèces (> 80 % de la radioactivité retrouvée) et dans une moindre mesure dans l'urine (≤ 19 %). Moins de 0,1 % de la dose excrétée a été retrouvée en tant que composé parent dans les urines.
Interactions médicamenteuses
Effets du trametinib sur les enzymes métabolisant et transportant les médicaments : les données in vitro et in vivo suggèrent qu'il est peu probable que le trametinib affecte la pharmacocinétique d'autres médicaments. Sur la base d'études in vitro, le trametinib n'est pas un inhibiteur de CYP1A2, CYP2A6, CYP2B6, CYP2D6 et CYP3A4. In vitro, le trametinib s'est avéré être un inhibiteur de CYP2C8, CYP2C9 et CYP2C19, un inducteur de CYP3A4 et un inhibiteur des transporteurs OAT1, OAT3, OCT2, MATE1, OATP1B1, OATP1B3, Pgp et BCRP. Cependant, sur la base de la faible dose et de la faible exposition systémique clinique par rapport à la puissance des valeurs inhibitrices et inductrices in vitro, trametinib n'est pas considéré comme un inhibiteur ou inducteur in vivo de ces enzymes ou transporteurs, bien qu'une inhibition transitoire des substrats des BCRP puisse survenir au niveau intestinal (voir la rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Effets des autres médicaments sur le trametinib : les données in vivo et in vitro suggèrent qu'il est peu probable que la pharmacocinétique du trametinib soit affectée par d'autres médicaments. Le trametinib n'est pas un substrat des enzymes CYP, ni des transporteurs BCRP, OATP1B1, OATP1B3, OATP2B1, OCT1, MRP2, et MATE1. Trametinib est un substrat de BSEP et du transporteur d'efflux P-gp in vitro. Bien qu'il soit peu probable que l'exposition au trametinib soit affectée par l'inhibition de BSEP, une augmentation des taux de trametinib secondaire à une puissante inhibition du P-gp hépatique ne peut être exclue (voir la rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Effets du trametinib sur d'autres médicaments : l'effet d'une dose répétée de trametinib sur l'état d'équilibre pharmacocinétique de contraceptifs oraux combinés, norethistérone et ethinylestradiol, a été évalué dans une étude clinique comprenant 19 patientes avec des tumeurs solides. L'exposition à la norethistérone a été augmentée de 20% et celle à l'ethinylestradiol est restée similaire lorsqu'ils étaient administrés concomitamment au trametinib. Sur la base de ces résultats, aucune perte d'efficacité des contraceptifs hormonaux n'est attendue lorsqu'ils sont administrés concomitamment au trametinib.
Populations particulières
Insuffisance hépatique
Les analyses pharmacocinétiques de la population et les données issues d'une étude clinique de pharmacologie chez des patients adultes ayant une fonction hépatique normale ou une élévation légère, modérée ou sévère des taux de bilirubine et/ou d'ASAT (selon la classification du National Cancer Institute [NCI]) indiquent que la fonction hépatique n'a pas d'effet significatif sur la clairance orale du trametinib.
Insuffisance rénale
Il est peu probable que l'insuffisance rénale ait un effet cliniquement pertinent sur la pharmacocinétique du trametinib du fait de la faible élimination rénale du trametinib. La pharmacocinétique du trametinib a été caractérisée par une analyse pharmacocinétique de population, chez 223 patients adultes qui présentaient une insuffisance rénale légère et 35 patients adultes qui présentaient une insuffisance rénale modérée inclus dans les essais cliniques avec le trametinib. L'insuffisance rénale légère à modérée n'a pas d'effet sur l'exposition au trametinib (< 6 %). Aucune donnée n'est disponible chez des sujets présentant une insuffisance rénale sévère (voir la rubrique Posologie et mode d'administration).
Origine ethnique
Les données sont insuffisantes pour évaluer l'effet potentiel de la race sur les paramètres pharmacocinétiques du trametinib dans la mesure où l'expérience clinique est limitée aux patients caucasiens.
Genre
Sur la base d'analyses pharmacocinétiques de population chez les patients adultes et pédiatriques, il a été constaté que le sexe influençait la clairance orale du trametinib. Bien qu'il soit prévu que les patientes soient plus exposées que les patients masculins, ces différences sont peu susceptibles d'être cliniquement pertinentes et aucun ajustement posologique n'est justifié.
Le trametinib a une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines. L'état clinique du patient et le profil des effets indésirables du trametinib doivent être pris en compte lors de l'évaluation de la capacité du patient à effectuer des tâches qui font appel au discernement, à des aptitudes motrices et cognitives. Les patients devront être informés du risque potentiel de fatigue, de sensations vertigineuses ou de problèmes oculaires affectant ces activités.
Aucune étude de carcinogénicité n'a été réalisée avec le trametinib. Le trametinib n'était pas mutagène dans les tests évaluant les mutations sur des bactéries, les aberrations chromosomiques dans les cellules de mammifères en culture et dans un test conduit sur des micronoyaux dans la moelle osseuse chez des rats.
Le trametinib peut affecter la fertilité chez les femmes. Dans les études à doses répétées, une augmentation des follicules et une diminution des corps jaunes ovariens a été observée chez les rats femelles à des expositions inférieures à l'exposition clinique humaine, d'après l'ASC.
De plus, il a été observé chez de jeunes rats sous trametinib, une diminution du poids ovarien, un léger retard des caractéristiques de maturité sexuelle chez la femelle (ouverture vaginale et augmentation de l'incidence de bourgeons terminaux proéminents dans la glande mammaire) et une légère hypertrophie de la surface de l'épithélium de l'utérus. Tous ces effets étaient réversibles suite à une période d'arrêt du traitement et attribuables à la pharmacologie. Cependant, dans les études de toxicité chez le rat et le chien conduites sur 13 semaines au maximum, aucun effet du traitement n'a été observé sur les tissus reproductifs des mâles.
Dans les études de développement embryo-foetal chez les rats et les lapins, le trametinib a entraîné une toxicité maternelle et sur le développement embryonnaire. Chez les rats, une diminution du poids du foetus et une augmentation des pertes post-implantatoires ont été observées à des expositions inférieures ou légèrement supérieures aux expositions cliniques chez l'Homme, d'après l'ASC. Dans une étude de toxicité sur le développement embryo-foetal chez des lapines, une diminution du poids foetal, une augmentation des avortements, une augmentation de l'incidence d'ossification incomplète et de malformations du squelette ont été observées à des expositions inférieures aux expositions cliniques, d'après l'ASC.
Dans les études à doses répétées, les effets après exposition au trametinib sont principalement retrouvés au niveau de la peau, du tractus gastro-intestinal, du système hématologique, des os et du foie. La plupart des cas sont réversibles après disparition du médicament. Chez les rats, une nécrose hépatocellulaire et des élévations des transaminases ont été vues après 8 semaines à des expositions ≥ 0,062 mg/kg/jour (environ 0,8 fois l'exposition clinique humaine, d'après l'ASC).
Chez la souris, un ralentissement cardiaque, une diminution du poids du coeur et de la fonction du ventricule gauche ont été observés sans histopathologie cardiaque après 3 semaines à des doses ≥ 0,25 mg/kg/jour de trametinib (environ 3 fois l'exposition clinique humaine, d'après l'ASC). Chez les rats adultes, une minéralisation de différents organes a été associée à une augmentation de la phosphorémie et a été étroitement liée à une nécrose du coeur, du foie et des reins, une hémorragie pulmonaire à des expositions comparables à l'exposition clinique humaine. Chez le rat, une hypertrophie osseuse et une augmentation du renouvellement osseux ont été observées ; cependant cette hypertrophie osseuse n'est probablement pas cliniquement pertinente chez les adultes humains. Chez le rat et le chien recevant du trametinib à des doses égales ou inférieures aux doses cliniques chez l'Homme, une nécrose de la moelle osseuse, une atrophie lymphoïde du thymus et dans le tissu lymphoïde associé au tube digestif (GALT) et une nécrose lymphoïde des ganglions lymphatiques, la rate et le thymus ont été observées, pouvant affecter la fonction immunitaire. Chez les jeunes rats, une augmentation du poids du coeur sans histopathologie a été observée à une dose de 0,35 mg/kg/jour (environ deux fois l'exposition clinique chez l'Homme sur la base de l'ASC).
Trametinib s'est révélé phototoxique lors d'un test in vitro 3T3 NRU (Neutral Red Uptake) réalisé sur fibroblastes de souris à des concentrations significativement supérieures aux expositions cliniques (CI50 de 2,92 µg/mL, ≥ 130 fois l'exposition clinique chez l'Homme estimée sur la base de la Cmax), suggérant que le risque de phototoxicité chez les patients prenant trametinib est faible.
Association au dabrafenib
Au cours d'une étude chez le chien, dans laquelle trametinib et dabrafenib ont été administrés en association pendant 4 semaines, des signes de toxicités gastro-intestinales et une diminution de la cellularité lymphoïde du thymus ont été observés à une exposition plus faible que chez les chiens recevant trametinib seul. Les autres toxicités étaient similaires à celles observées dans les études comparables en monothérapie.
La solution buvable de Trametinib est préférentiellement à faire reconstituer par un pharmacien avant la délivrance
Instructions pour la reconstitution : Veuillez vous référer au guide de reconstitution à destination des personnes en charge du traitement Trametinib en solution buvable.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Médicament soumis à prescription hospitalière.
Médicament à
prescription réservée aux spécialistes en oncologie ou aux médecins compétents
en cancérologie.
Médicament nécessitant une surveillance particulière pendant le traitement.
Poudre pour solution buvable.
Poudre blanche ou blanchâtre.
Flacon en verre ambré avec un bouchon équipé d'une sécurité enfant.
Chaque boîte contient un adaptateur pour le flacon (PIBA).
Chaque flacon contient du diméthylsulfoxyde de trametinib correspondant à 4,7 mg de trametinib. Chaque ml de la solution reconstituée contient 0,05 mg de trametinib.
Excipients à effet notoire
Chaque ml de solution reconstituée contient 100 mg de sulfobutyle bétadex de sodium, 0,8 mg de méthyle de parahydroxybenzoate et 1,98 mg de sodium.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Sulfobutyle bétadex de sodium
Sucralose (E955)
Acide citrique monohydraté (E330)
Phosphate de sodium (E339)
Sorbate de potassium (E202)
Parahydroxybenzoate de méthyle (E218)
Arôme fraise